

A peek inside AlphaFold: The deep learning model that fetched a Nobel Prize

Introduction
This year, 2024, John Jumper is going to get the Nobel Prize in chemistry, due to an impressive feat of designing a neural network model called “AlphaFold2”, which is extremely accurate in predicting new protein structures. It is even more impressive that he is just 39 years old, almost two decades younger than the average age of 61 years of the Nobel Prize winners.
In this post, I am going to try to provide a sneak peek into the problem that “AlphaFold2” solves, and how it solves it. Since I have almost zero background on biology and chemistry, I will try to explain it in very “layman” terms — which is clearly not doing proper justice to the work done by Dr. Jumper and his many coauthors at Deepmind — but an interested reader may always read the original papers to get more technical details, which I will cite in the post as appropriate.
What is protein folding prediction problem?
Even before that…
What is a protein anyway?
Protein is the building block of life. All live things, if you break them down enough at the sub-cellular level, it is always a blob of protein — a special kind of chemical that is essential for life to thrive.
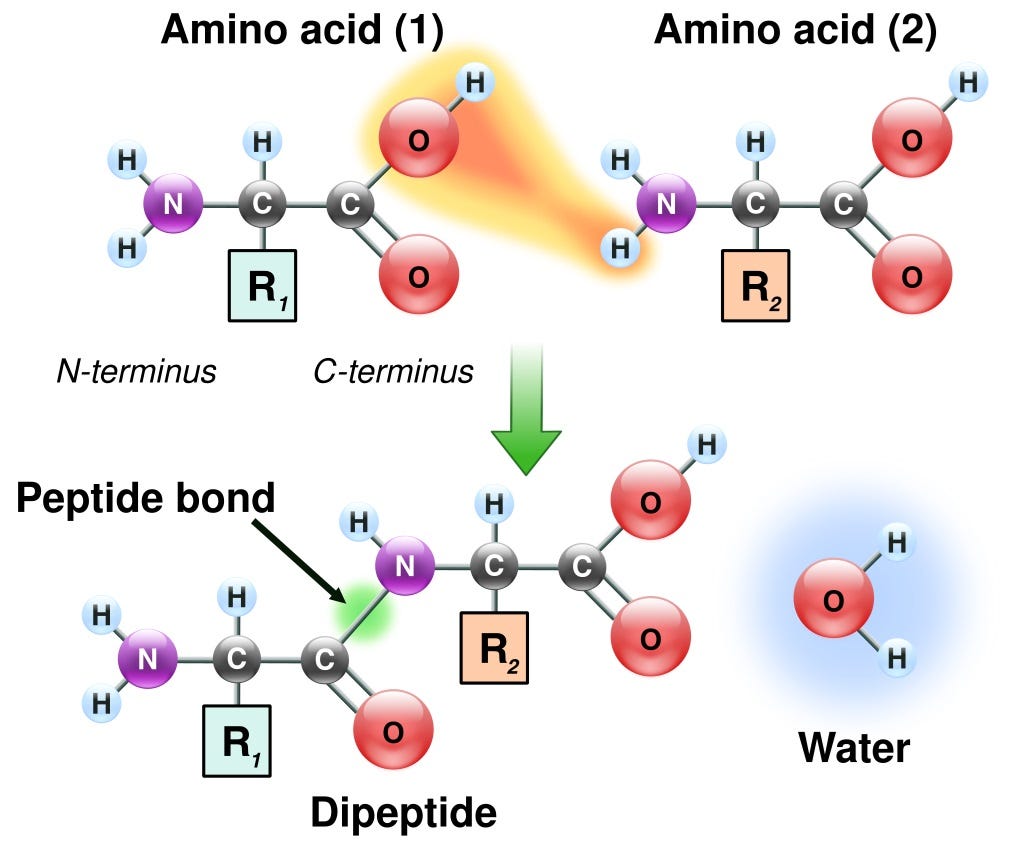
If you break down a protein, you will see it is like a chain of amino acids. All amino acids share a basic structure.
You have a central carbon atom, called the 𝛼-carbon. It has 4 hands connected to 4 components.
This carbon atom is bound to an amino group or NH2 (2 Hydrogen atoms + 1 nitrogen atom)
On the other side, this carbon atom is bound to a carboxyl group (COOH). The carbon atom in this group is called 𝛽-carbon.
On another side, the 𝛼-carbon is connected to a hydrogen atom.
Finally, this 𝛼-carbon is also attached to a residual group (denoted by R), which can vary depending on the type of the amino acid. This residual group determines the chemical properties of the entire amino acid.
There are only 20 types of amino acids that govern almost all of life.

Now once you have a bunch of amino acids lying around, some of them can come close to each other and start having some sort of chemical reactions. Due to this reaction, some of them bond together and create a 3-dimensional structure. This 3D structure is called a “protein”.
To understand how this bond is created, it is usually simple. One amino acid’s amino group has 2 hydrogen atoms, and the carboxyl group has an oxygen atom. Then gets connected together to produce H2O (or water) and the other things properly join together into a bond called a “peptide” bond.
3d structure of a protein
Now to make a protein structure from a chain of amino acid residues (the amino acid “block” or “group” that we talked about), for each residue, it can connect to any other residue based on its position and rotation. However, there are two fundamental physical laws it should satisfy:
(1) The structure needs to make a closed loop. If there are certain parts of the structure which is poking out as a rod, it is easy to get broken due to external forces.
(2) The ultimate configuration for the structure of the protein is the one with minimal free energy among many valid configurations satisfying (1).
For instance, this is the 3D structure for human haemoglobin (ID: 1GZX)

This is a nice website where you can view the same https://www.rcsb.org/3d-view/
Protein Folding
Now it turns out that, if you start with a chain of amino acids on a viscous solution, nature is so “intelligent” that it solves the minimization of the free energy problem itself, and the chain of the acid automatically gets folded into a 3D structure.
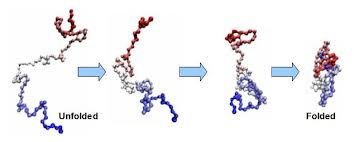
And surprisingly, if you start with two identical copies of amino acid chains, they both end up having the same folded 3d structure. Thus, the chain itself describes what kind of protein structure we will end up with —
Conversely, to know the output protein, it is actually enough to know the unfolded sequence of amino acids we are starting with.
But it is important to know the 3d structure of the protein because only that structure allows us to derive its physical, biological and chemical properties.
If a sequence of amino acids does not fold into a 3D structure, it remains inactive, and it cannot be a building block for any life.
This process is called “Protein folding”.
This discovery was so intriguing, that Christopher Boehmer Anfinsen, a renowned biochemist, won the Nobel Prize for this work in 1972. He proved by a series of experiments that “All the information for the native fold (the 3D structure on the right end of the image) appears, therefore, to be contained within the primary structure (the unfolded chain on the left end of the image).”
Why predicting protein folding is hard?
In addition to the 2 fundamental laws for protein folding described above, there are many other variables involved in the folding process:
Minimizing the number of hydrophobic side-chains exposed to water (the hydrophobic effect) is an important driving force behind the folding process.
Intramolecular hydrogen bonds also contribute to protein stability (think of their importance in secondary structures).
Ionic interactions (attraction between unlike electric charges of ionized R-groups) also contribute to the stability of tertiary structures.
Disulfide bridges (covalent bonds) between neighbouring cysteine residues can also stabilize three-dimensional structures.
To me, they are like blah, blah, blah … the point I am trying to make here is that the protein folding process has tons of variables so it is difficult to track them all.
Although, nature can solve this automatically for us, physically creating the chain of amino acids and observing the folding process unfold, is time-consuming, and erroneous. Also, it is extremely difficult to build a long unstable chain of amino acids in the first place.
The other solution is to use physics or chemical simulator software to perform the folding process inside a computer, which may take days or even months. Usually these software consider many valid configurations of the 3d structure, and see which one yields the minimal free energy. Consider the following naive calculation: Suppose you have 50 amino acids, each acid has 2 rotatable bond groups (NH2 and COOH), so 100 groups. Each group in a 3D structure can rotate into different angles, suppose we consider only angles in 10-degree increments, so that becomes 36 angles for the whole 360 degree. Together we have 100³⁶ possibilities. Even if some of them are equivalent due to rotational symmetry, it is still a very big number, well beyond the number of atoms in the universe.
Btw, a chain of length 50 is actually small. The median length of the human protein is 375, i.e., 7.5 times. See this article for details.
AlphaFold
Alphafold, being a deep learning system, solves this particular problem. You give it a chain of amino acids, it predicts the 3D structure of the protein. Then you can analyze the 3D structure and get all the necessary properties of the protein.
For example, you want to get a cure for Sars-Cov2 (COVID) virus? Sure! Come up with a bunch of sequences of amino acid residues that you think will work. Use AlphaFold to predict the 3D structures. And then, analyze those 3D structures to see if any of them has the properties to cure COVID.
If any of them are, good, you start doing physical experiments.
If none of them are good enough, well, start again with some more guesses. Or better yet, since you are just predicting sequences of letters (each amino acid is usually denoted by a group of letters representing its ID), you can use LLMs like ChatGPT to generate this! 😎
Okay, now that we understand a little bit about what AlphaFold does, we can see how it solves the problem. But before that, AlphaFold actually has 2 versions. The second version won the Nobel Prize, but we need to talk about the first version to understand things better.
AlphaFold Version 1
CASP (Critical Assessment of Structure Prediction) is a community-wide experiment to determine the state of the art in modelling protein structure from amino acid sequences organized by the Protein Structure Prediction Center. It also hosts a competition in which participants submit models that attempt to predict a set of proteins for which the experimental structures are not yet public.
AlphaFold from Deepmind participated in the CASP13 competition in 2018, where it won the prize. It achieved a summed z-score of 68.3 compared to 48.2 for the next best method. That’s nearly a 40% improvement. It was at that time that people started to believe that deep neural networks have the potential to solve this problem.
AlphaFold had 2 stages in its architecture. The first stage is a convolutional neural network. The second stage is a simple gradient descent step for minimization of a potential function.
For a given sequence, they first search over a training dataset to find similar protein sequence, and produce a multiple sequence alignment (MSA) of the target sequence. This training dataset is often available from the Protein Data Bank. Think of MSA as a sophisticated searching mechanism, which can match a target sequence of residues to a substring of a sequence present in the database and vice-versa.
For each pair of residues i and j, in total 485 MSA features were extracted and provided as input to the model.
So the input was a tensor of size L * L * 485, where L is the length of the chain.
Then they has a series of convolutional blocks, in particular 220 blocks of 64 * 64 kernel size. This convolutional neural network had 7 groups of 4 blocks with 256 channels, cycling through dilations 1, 2, 4, 8, and also 48 groups of 4 blocks with 128 channels, cycling through dilations 1, 2, 4, 8. They also added some batch normalization layers.
After processing through this neural network, the output was a tensor of size L * L * 64. For each pair of residues i and j, the 64 channels described the probability of the distances between the central C𝛽 atoms of residue i and residue j, over 64 different bins. The bins represented distances from 2 angstroms to 22 angstroms.
This structure is often called a “distogram”, or distance histogram.
Once the distogram is computed, the next step is to actually compute the 3D structure. To draw the 3d structure, it is often enough to know the angles at which each residue will be rotated. Denoting the angles as 𝜙, 𝜓 (think of azimuth and zenith angles when you view the sky), one can then write the distances as a function of these angles.
So now you have the distances coming from the first step of the network.
And you have the distances as a function of the 3d angles of rotation.
So, they write an objective function regarding the dissimilarity between these two, and then minimize the objective using a gradient descent approach.
The final solution will give the angles, hence the 3d structure of the protein.
This is the network architecture diagram taken directly from the AlphaFold paper, for more details about the network, please see the original paper.

Problems with version 1
There were two major problems with AlphaFold version 1, despite its groundbreaking accuracy.
It uses a convolutional neural network that takes only local affinities. However, it is possible that residues on either end of sequences may come in contact and form a bond, because of rotational movements (also as it needs to make a closed loop).
Another possible drawback of AlphaFold is choosing the distogram as the main representation of a 3d structure. The space of possible protein foldings is very small compared to the space of possible distograms. Because the network isn't constrained in any way, it can output distograms that are impossible in the real world (for example, a distogram that does not satisfy the triangle inequality).
AlphaFold Version 2
AlphaFold2 was the winner of the CASP14 competition. It achieved an impressive summed z score of 244.0217, the closest competitor achieved a summed z score of 92.1241, which clearly shows a great improvement over the existing methods.
AlphaFold2 has results outside the CASP competition as well. AlphaFold2 predictions have been validated against the structure of ORF3a where the results have ended up being similar to structures later determined experimentally despite their challenging nature and having very few related sequences, see this paper by Flower and Hurley, 2021.
AlphaFold2 has some novel changes compared to its predecessor.
The AlphaFold network directly predicts the 3D coordinates of all heavy atoms for a given protein using the primary input sequence and MSA features, instead of going through a distogram route.
In the training procedure, it added some constraints based on physical and geometrical properties to the 3d structure. It made sure that the network does not predict protein structures that are impossible in the real world.
AlphaFold2 architecture contains again 2 stages:

An evoformer block: This is a new block that has various attention type and non-attention type blocks.
Based on the input sequence, it searches over the training data to find related MSA sequences. Instead of extracting different MSA features, the raw MSA features (n * L) is fed into the network. Here, n is the number of MSA sequences retrieved.
As an output of this network block, we obtain two things:
A processed MSA sequence of size n * L.
A processed correlation structure of size L * L * c, where for each pair of residues i and j, we obtain c different features for representing their correlation.
Inside this block, the key idea is to come up with features of a network graph, where the edges of the graph are defined by residues in proximity (i.e., they come in close contact and join by a peptide bond).
Because a graph is used, it automatically satisfies the rotational and translation invariance, and rules out geometrically equivalent structures.
Once the graph was constructed from the pairwise correlation representation, to satisfy the triangle inequality, they identified all triangles in the graph. Then the corresponding edge weights are updated by a multiplication of the other two edge weights of the triangle.

These two outputs are then fed into a structure module. This module outputs the 3D coordinates of the protein structure.
The parameters of this structure module are 2 things for each residue. One, a rotation parameter and another, a translation parameter. During initialization, the rotation is set to identity and the translation is set to zero.
They use an iterative refinement technique here. Once the output 3d structure is obtained, based on the proximity of C𝛼 atoms, a neighbour graph is constructed and a correlation tensor is computed based on the neighbour graph. Then that correlation tensor is again fed to the same network. This iterative method, which is repeated 3 times, allows the same network to reason about better structures and perform local adjustments if necessary.
However, though this controls the geometrical 3D structure, this does not have any chemistry constraints. So, it may violate some peptide bond constraints, as breaking this constraint enables the local refinement of all parts of the chain during the iterative refinement process.
Due to this, the authors tried to enforce the peptide bond geometry after the network is trained, as a post-training relaxation step. In this, they perform fine-tuning the network by adding an additional loss term which is large if chemical constraints are violated.
Interested nerds can find more details in the original paper.
Conclusion
Well, the story does not end here. In 2024, we have yet another improved version of AlphaFold.
AlphaFold changed the biological landscape for understanding protein structure in a better way. The process which used to take months and years, it can now help us do that in minutes, and that too with a remarkable degree of accuracy. AlphaFold has found its application over hundreds of use-cases, impacting millions of people. Some of these use cases are showcased on Google Deepmind’s website.
Also, the AlphaFold database and its server are accessible from any computer, with an internet connection. So essentially, you have the best protein prediction tool available out there at your fingertips, and this means, for the next disease outbreak like COVID, we all can be prepared for it, and maybe, find a cure on our own!
Thank you very much for being a valued reader! 🙏🏽 Subscribe below for more stories about scientific breakthroughs that changed the world! 📢
Until next time.